 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipDisulfide rich peptides are being identified in species of both plants and animals at increasing rates. As new molecules are discovered and disulfide bonding patterns characterized, the need for simplified chemical synthesis strategies is also increasing.
I have previously written about optimizing removal of several orthogonal side chain protecting groups including allyl, alloc, ivDde and acetamidomethyl (Acm) groups. The question that I’ll address today, though, is does the order in which the disulfide bonds are formed matter for cleaning up reactions to produce chemically synthesized disulfide rich peptides?
There have been many strategies published which enable cysteine oxidation to cystine, both in solution and while the peptide is still on resin. As the complexity of peptides increases, these strategies require much more careful consideration and several preliminary questions come to mind:
- Which orthogonal protecting groups will be used for on-resin disulfide bonds?
-
Is there a particular order for disulfide bond formation that will lead to the most success in purification?
The principle advantage to chemical synthesis using on-resin disulfide bond formation for these peptides is the regiospecific control that can be maintained. This preserves the specific disulfide bonding pattern critical to the structure and function of these peptides while decreasing the potential purification burden.
To address the second question, I’ve synthesized apamin, an 18 amino acid peptide containing 4 cysteines joined by 2 disulfide bonds originally isolated from bee venom. I first synthesized the linear peptide sequence using my Biotage® Initiator+ Alstra™ to identify any regions of the sequence that could pose synthetic challenges, Figure 1.
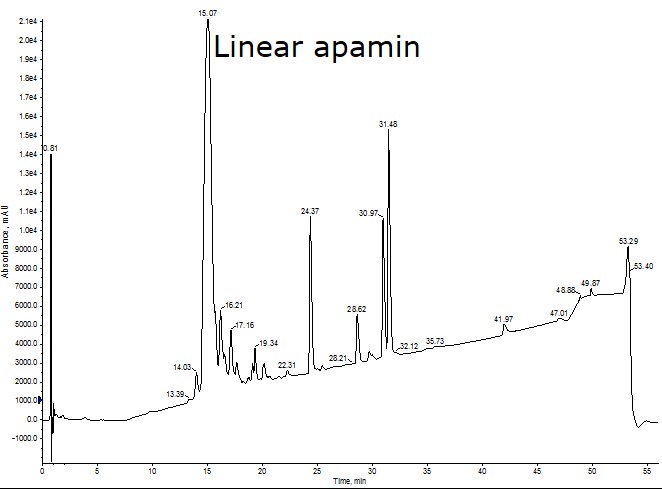
Figure 1: Crude analytical HPLC of linear apamin. The high intensity, late eluting peaks contain no peptidic character when analyzed by mass spectrometry and are likely attributed to the presence of residual protecting groups in the solution. The crude peptide was synthesized in 81% crude purity when the protecting group peaks are not counted in the total absorbance.
The linear sequence, although rich in both cysteine and arginine, is relatively straightforward to synthesize with high crude purity (81%). With a synthetic strategy for the linear sequence in hand, I decided to use cysteine residues protected with 4-methoxytrityl (Mmt) and Acm protecting groups for the on-resin disulfide bond formation. I decided on these protecting groups for two reasons: 1) there is sufficient orthogonality for both the deprotection and cysteine oxidation conditions, and 2) I have worked with each individually minimizing the degree of optimization that will be required for this specific peptide. It is important to note that iodine-mediated Acm removal can also lead to removal of the trityl and Mmt protecting groups, so this must be used for the second oxidation step to ensure the proper disulfide bonding pattern.
I utilized the Branches™ software feature of the Biotage® Initiator+ Alstra™ to simplify programming the specific disulfide bonding pattern and the associated chemisty, Figure 2.
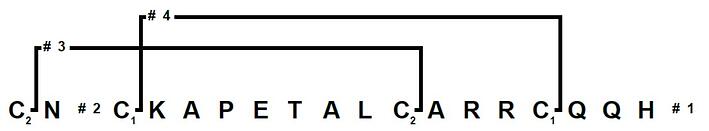
Figure 2: Branches™ representation of apamin for fully automated synthesis and cysteine oxidation. Each cysteine subscript represents an orthogonally protected cysteine residue while the numbers dictate which order the synthesis will occur.
Biologically relevant apamin contains disulfide bonds between Cys 1 and 11 as well as between Cys 3 and 15. Given this configuration, I decided to form the internal disulfide (3-15) using an Mmt protected cysteine pair first and the N-terminal disulfide (1-11) using an Acm protected cysteine pair second, Figure 3.
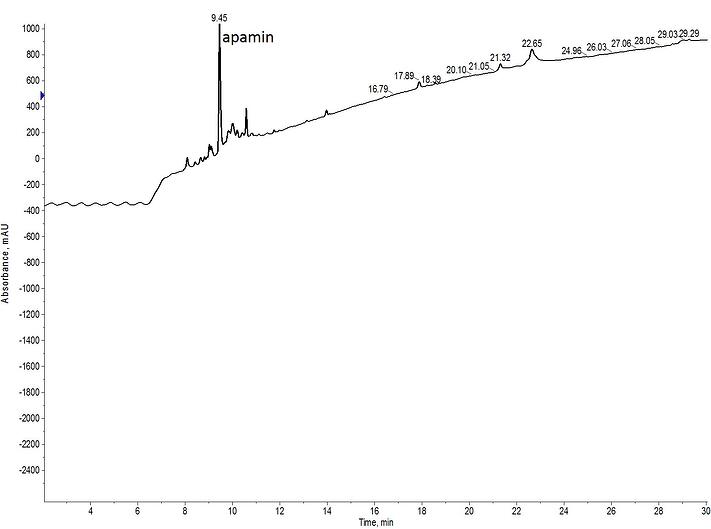
Figure 3: Crude analytical HPLC of fully oxidized apamin, synthesized by forming the internal disulfide bond (3-15) first and the N-terminal disulfide bond (1-11) second.
This approach worked very well! The sharpness of the peak suggests that the peptide is adopting a single conformation and both disulfide bonds fully formed. Importantly though, the efficiency of the linear sequence is maintained through the additional chemistry and I would expect a simplied purification given the purity of this crude peptide sample.
The question though is does this efficiency still apply if the disulfide bonds are formed in the reverse order? Using the same deprotection and oxidation protocols, I synthesized apamin with Mmt protected cysteine at positions 1 and 11 and Acm protected cysteine at positions 3 and 15, Figure 4.
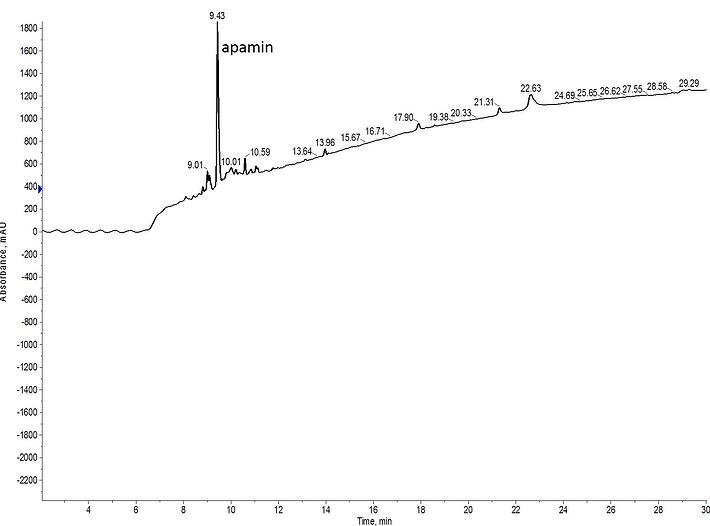
Figure 4: Crude analytical HPLC of fully oxidized apamin, synthesized by forming the N-terminal disulfide bond (1-11) first and the internal disulfide bond (3-15) second.
I was concerned that the steric bulk and reduced conformational accessibility caused by the presence of the N-terminal Fmoc protecting group would limit the success of this approach. But this appears to not be the case. As with the first synthesis, a nice sharp peak is observed in the crude chromatography with the same retention time, suggesting a single peptide conformation state with both disulfide bonds formed stably .
So why was this so effective? There is certainly evidence that not all disulfide bonds are created equal, demanding a specific order for disulfide bond formation to achieve the highest crude purity and yield. Accessible conformational flexibility, distance in linear sequence and the identity of the intervening amino acids can all play a role in the kinetic efficiency of the oxidation reaction. In the case of apamin, there seems to be sufficient conformational flexibility and separation in linear sequence to allow for either of the disulfide bonds to efficiently form first. Importantly for this peptide, formation of the first disulfide bond likely drives a conformation that positions the remaining two cysteines in close proximity, enhancing the probability for a successful second oxidation step.
It would be naïve to suggest that this will be the case for all disulfide rich peptides. I will certainly be exploring the rigidity of these observations for more complex peptides in the future and I look forward to learning if this is actually more general than I anticipate.
What is your favorite strategy for forming multiple disulfide bonds while the peptide is still on the resin?
If you would like to learn more about how Biotage® tools can simplify your peptide workflow, follow the link below.