 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipSide reactions. Words that cause a little shiver to run down every peptide chemists’ spine. As peptide chemists, we worry about both chemical side reactions like diketopiperazine or aspartimide rearrangements, and secondary structure formation as causes for failed peptide syntheses. But how do you know what to look for? What is a susceptible sequence and how can you confirm if one of these structural rearrangements has occurred?
In today’s post, I’ll discuss a couple strategies that have been published that illustrate how to identify if an aspartimide rearrangement has in fact occurred during your peptide synthesis.
As a young graduate student, I ran into a problem. I was working with a peptide that displayed batch-to-batch variations in activity when evaluated against a receptor over-expressed on the surface of a mammalian cell, Figure 1. There was no significant retention time shift in my analytical HPLC after purification and I only saw the desired peptide mass. So what was going on?
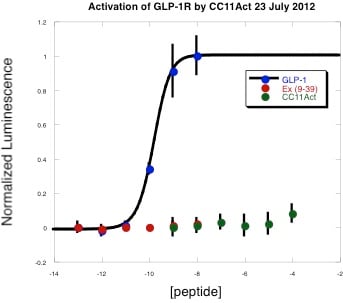
Figure 1: GLP-1R is potently activated by its cognate ligand GLP-1 (blue circles), not the antagonist exendin-4 (9-39) (red circles) and not by CC11Act (green circles), a synthetic peptide mimetic that had previously stimulated receptor activation.
After a bit of reading, I began to suspect that my peptide had undergone an aspartimide rearrangement, inserting an isoAsp bond into the peptide causing a reduction in the peptide’s overall efficacy against the receptor of interest, Scheme 1. This is a well known isomerization reaction, documented as early as 1979, that has a sequence-dependent probability of occurring during synthesis.
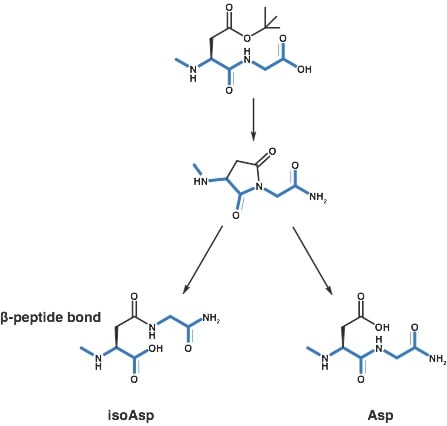
Scheme 1: Formation of an isoAsp bond through a succinimidyl intermediate during Fmoc-based peptide synthesis.
IsoAsp is particularly difficult to detect in peptides for 2 key reasons:
- -The probability that the rearranged product will be resolved from the desired peptide chromatographically is very small (this is similar to separating small molecule enantiomers).
- -The rearrangement is mass neutral, especially if using an ESI ionization source in the mass detector.
What I needed was a quick and reliable method to characterize each synthesis batch of this peptide to determine if in fact this an isoAsp was present in the peptide. Ideally, the method would be relatively quick, reliable, and HPLC-MS compatible for analysis. The literature presented several strategies including the use of enzymes and HPLC-MS, enzymes with both heavy and radio isotopes, or alternative ionization methods. Below is a brief summary of some useful strategies for detecting and quantifying isoAsp in peptides and protein samples, Table 1.
Method for Detecting isoAsp Formation
| Strategy | Summary | Pros | Cons |
|---|---|---|---|
| S-[methly-3H]AdoMet and PIMT* | PIMT selectively transfers active ester on AdoMet to free side chain carboxylic acid, follow up hydrolysis releases labeled MeOH for quantification | sensitive quantification, reproducible (you can buy a kit now), | two-step peptide modification process, requires radioisotope |
| Asp-N endoproteinase digestion | Asp-N selectively cleaves the N-terminal side of Asp residues, leaving isoAsp containing peptides intact | selective for Asp-containing peptides only, no additional sequence constraints | desired material destroyed, |
| Electron capture dissociation mass spectrometry | peptide fragmentation strategy where low-energy electrons react with multiply-charged peptide species causing fragmentation | detectible mass difference in frgments generated by isoAsp and Asp containing peptides | requires specialized instrumentation |
| Edman degredation | process to chemically hydrolyze each amino acid from the peptide chain, reaction doesn't work at isoAsp | no enzymes necessary, non-isotopic | laborious, with significant time investment |
*This strategy has been employed using a variety of isotopes.
Ultimately, I worked out a method combining Asp-N endoproteinase digestion followed by UPLC-MS analysis. Although this strategy does cost sample (the desired peptide is destroyed in the digestion), I was able to complete a small scale digestion and follow up analysis in about 2 hours.
After working out the appropriate conditions for the digestion I circled back to the beginning and applied this additional level of characterization to the peptide sample shown above. As I suspected, the sample contained significant amounts of undigested peptide product, Figure 2, leading to the conclusion that the isoAsp-containing peptide caused the decrease is detectable peptide activity.
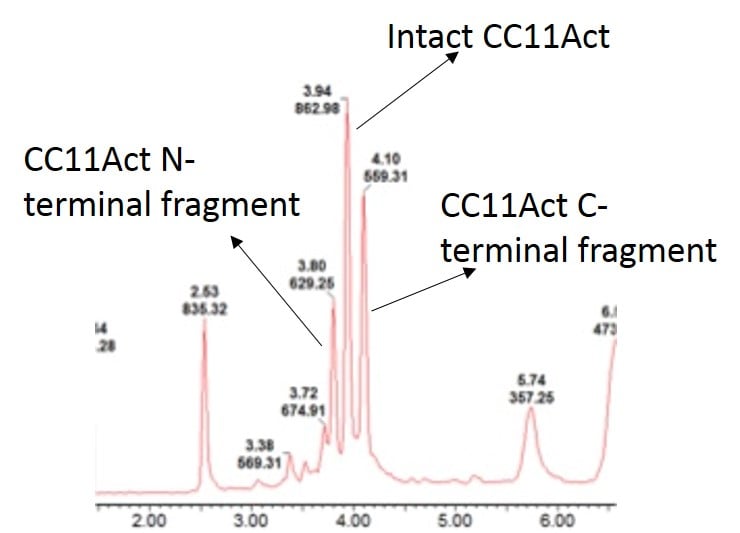
Figure 2: Digestion of CC11Act with Asp-N endoproteinase followed by UPLC-MS analysis. The sample contains both desired, Asp containing peptide and undesired, isoAsp containing peptide.
So now the question arises: how do I prevent this from happening in the first place? Stay tuned for that discussion in a future blog post.
To learn more about Biotage tools designed to improve your peptide workflow efficiency, click the link below.
Improving Peptide Workflow Efficiency