 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipAspartimide rearrangements are a particularly nasty side reaction that can occur during fmoc-based solid phase peptide synthesis. Not only is this a mass-neutral side reaction, chromatographically resolving the undesired, rearranged product can be particularly difficult. To make matters worse, this side reaction can occur at any point during the synthesis after the Asp has been incorporated into the peptide.
In a previous post, I described method that I have found useful for identifying whether or not an aspartimide rearrangement has occurred during synthesis of a peptide that contains an aspartimide-susceptible sequence. In today’s post, I’ll discuss some strategies that can be used to suppress, or even eliminate this side reaction.
Because an aspartimide rearrangement is so problematic, there has been a lot of work done of the years research different strategies to prevent the side reaction all together (check out this great review for more info).
One of the simplest strategies for limiting aspartimide formation is to change the Fmoc-removal conditions. Simply adding 0.1 M hydroxybenzotriazole (HOBt) to the piperidine solution has been shown to reduce aspartimide formation significantly. However, HOBt is an explosive substance when in its anhydrous state, and is exclusively sold today wetted, introducing nucleophilic water into the deprotection solution. Alternatively, piperizine, a weaker base, has been shown to be effective at removing the Fmoc group, while simultaneously suppressing aspartimide formation. It is important to note however, than neither of these methods eliminates the formation of rearranged products.
There has also been significant effort developing alternative protecting group strategies that can potentially eliminate this side reaction. One of the most obvious strategies is to develop a different protecting group for the Asp side chain carboxylic acid. Increasing the steric bulk of the side chain protecting group can effectively block succinimide ring formation, preventing the rearrangement. Many different protecting groups have been evaluated to help alleviate this problem. Some including, 3-methylpent-3-yl (Mpe), 2,3,4-trimethylpent-3-yl (Die), have shown improvements over t-butyl protected Asp while others, like Dmab, have actually shown an increase in aspartimide propensity. Interestingly, the protecting group needs to be not only bulky, but also somewhat flexible to ensure protection from the aspartimide rearrangement. Large, conformationally restrained protecting groups have shown little success.
The only method thus far shown to completely eliminate rearranged products is protection of the adjacent building block amine. Converting the standard, Fmoc-protected secondary amine to a tertiary amine removes the reactive lone pair of electrons. The Dmb protecting group is almost exclusively used at this point. Just like with proline, the secondary amine negatively impacts the coupling efficiency, so care must be taken when optimizing the incoming Asp amino acid coupling. To this end, amino acid suppliers routinely stock the Asp-Gly DMB-protected dipeptide, the most susceptible sequence, which can be readily incorporated into a growing peptide chain.
Each of these strategies comes with increased cost. The alternatively protected building blocks are more expensive than the standard Fmoc-Asp(OtBu)-OH, Table 1. But considering the alternative (think painful purification or repeated synthesis), the extra cost may be worth it.
| Name | Quantity (g) | Price (USD) | Structure |
|---|---|---|---|
| Fmoc-Asp (OtBu)-OH | 25 | $70.50 | 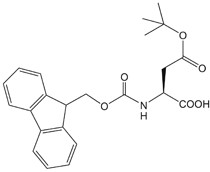 |
| Fmoc-Asp(Ompe)-OH | 1 | $267.00 | 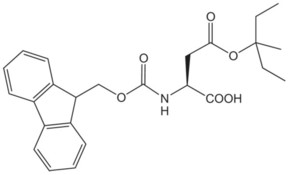 |
| Fmoc-(Dmb)Gly-OH | 1 | $295.00 | 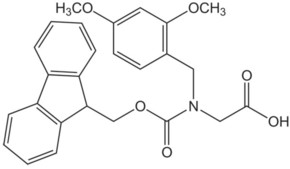 |
| Fmoc-Asp(OtBu)-(Dmb)Gly-OH | 1 | $305.00 | 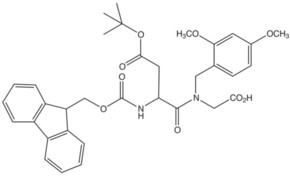 |
Table 1. Price comparison for a variety of Asp building blocks differing by protecting group strategy. Note: retail price listed from a single supplier.
For my past work, I found great success using the Asp(OMpe) protection strategy. My sequence contained an Asp residue adjacent to a Peg building block. This combination is reminiscent of the Asp-Gly sequence, and often yielded a significant amount of rearranged product. The Asp(OMpe) residue was readily incorporated into my peptides even when using a reduced number of equivalents, yielding peptides of much greater purity (and biological activity!).
Would you like to learn more about a quick strategy that can help to determine if the rearranged isoAsp is present in your sequence? Click the link below to learn more.