 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipIn the past, when I have synthesized a new peptide, I always ran a “scout run” – a small scale injection, usually with an analytical HPLC column – to both get an idea of the crude purity and also to identify a shorter, more optimal gradient for the actual purification. This strategy is still recommended when you want to use reversed phase flash chromatography for your purification strategy, but is there a better way?
In today’s post, I’ll discuss using a scouting column to screen gradient conditions prior to peptide purification with reversed phase flash chromatography.
Small molecule chemists have the luxury of using TLC to pre-screen conditions that will yield a more optimal purification. While reversed phase TLC plates are commercially available, they are typically unreliable for capturing the elution profile of your compounds of interest. This is primarily because the partitioning mechanism that occurs is simply not as efficient on the TLC plate surface. So what strategy can we use to predict our elution profile?
Typically, I run an analytical HPLC sample of my crude peptide and build my flash gradient around the acetonitrile concentration at which my peptide elutes. For example, in Figure 1, I evaluated a sample containing approximately equimolar quantities of a 17- and 18-amino acid peptide. Using the traditional analytical column, they are pretty well resolved and I calculated an approximate elution concentration of 16-18% acetonitrile.
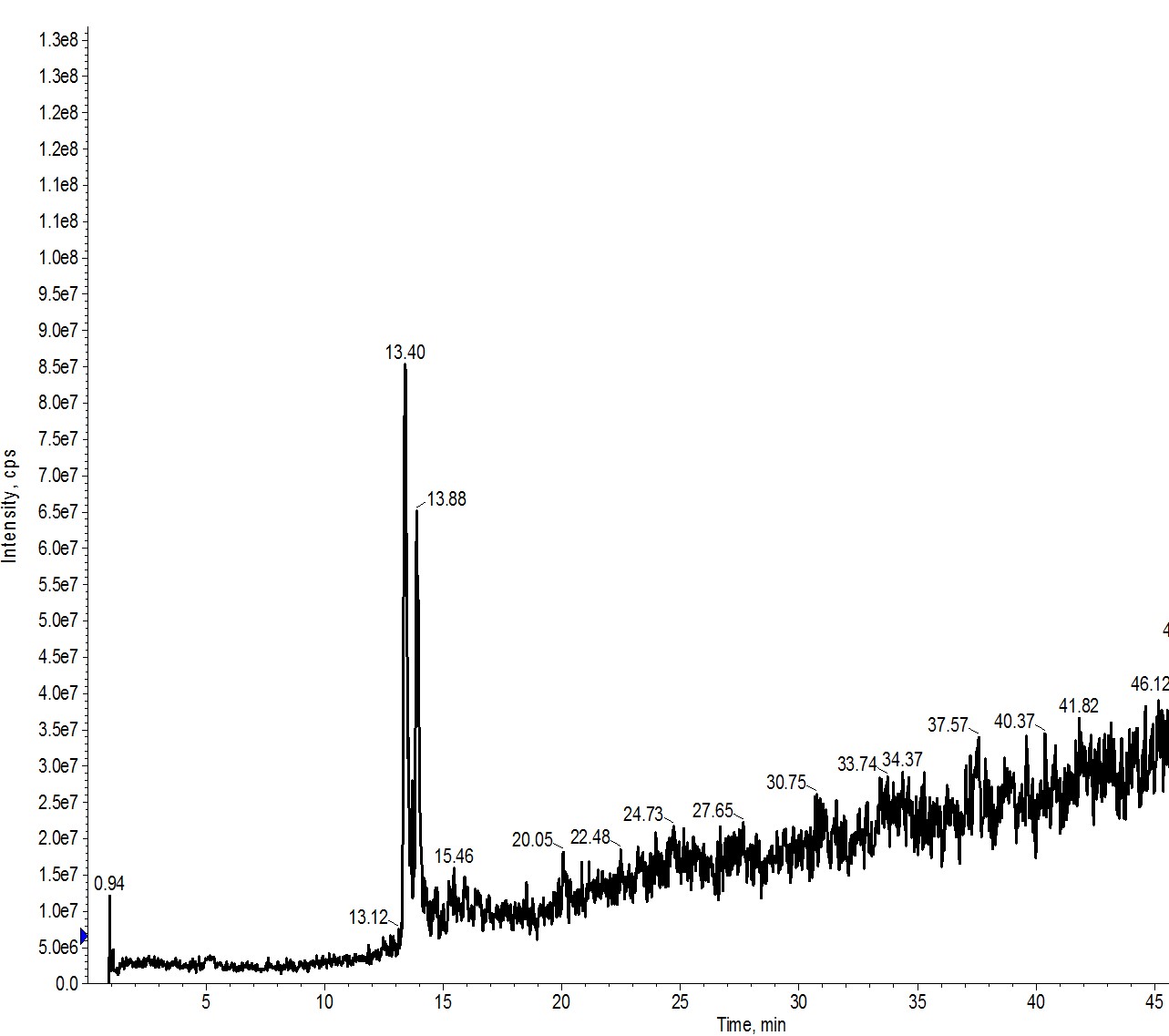
Figure 1: Analytical evaluation of a peptide sample containing a 17- (left peak) and 18-amino acid (right peak) peptide. The evaluation was completed using an analytical HPLC column (4.5 μm, 300 Å pore).
Using this information, I built my gradient for purification covering 0-50% MeCN over 10 column volumes. I have already discussed the impact that particle size and pore size have on resolution and purification efficiency, and sometimes this quick profiling leaves something to be desired when converting my HPLC gradient to a flash gradient, Figure 2.
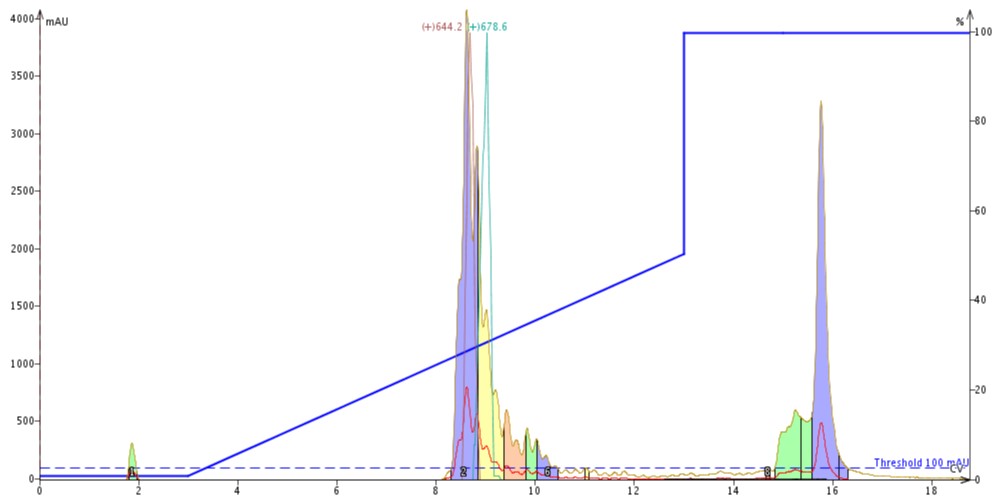
Figure 2: Crude peptide sample purified using a Biotage Isolera Dalton 2000 equipped with 10 g Biotage SNAP Bio C18 cartridge. The 17-amino peptide elutes early, as indicated by the 644.2 XIC trace (red) whereas the 18-amino acid peptide coelutes on the tailing side, as indicated by the 678.4 XIC trace (teal). The peptide elute approximately 30% MeCN, significantly later than predicted with the analytical HPLC.
Not only is the resolution severely compromised (sort of expected given the different particle sizes), but the elution concentration is also significantly different than I would have anticipated from the analytical HPLC. With such dramatic differences between the analytical outcome and the actual observed chromatographic behavior, it is difficult to have confidence that the purification will work well in the first attempt.
Alternatively, I could use a scouting column for the initial screening. A scouting column looks and operates just like a standard analytical HPLC column connected to a standard HPLC system. Rather than packing with traditional HPLC stationary phase (4.5, 3.0 or 1.5 μm particles, various pore sizes) though, a scouting column is packed with the same stationary phase as that used in flash chromatography (20 μm particles, 300 Å pore).
I first re-ran my crude peptide sample on my BioC18 scouting column using the same injection volume and purification gradient (0 to 80% MeCN over 40 min) as that used with my standard analytical HPLC column, Figure 3.
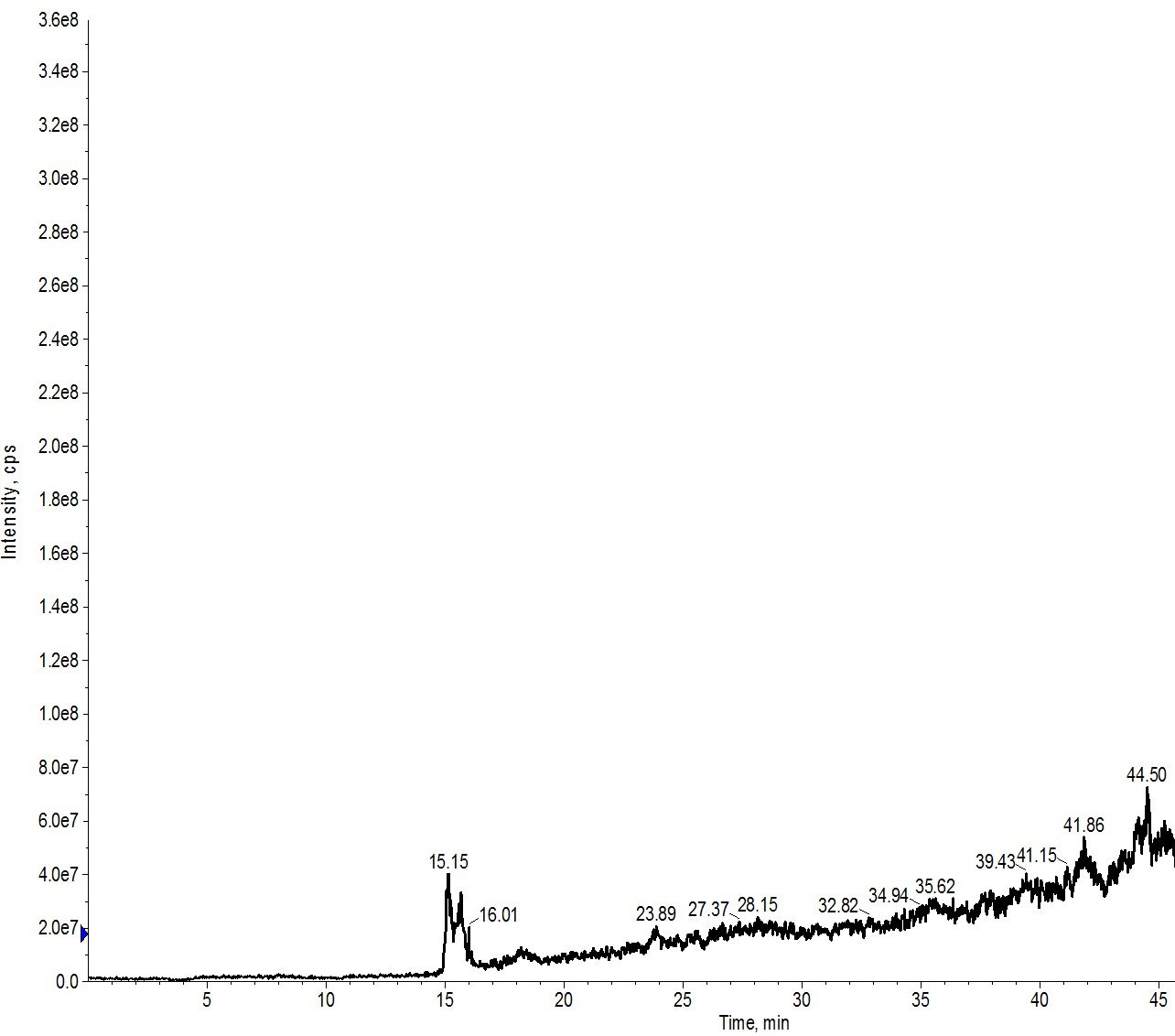
Figure 3: Crude peptide mixture evaluated using a BioC18 scout column. The sample was evaluated using a 0 to 80% MeCN gradient over 40 min.
Two key observations here. The first is that the peak to peak resolution has gone down a bit when compared to the analytical HPLC column. This isn’t very surprising though given the stationary phase is composed of a 20 μm particle. The peptides also elute slightly later than previously, but not nearly as late as what I observed in the purification run.
When I repeated the injection, this time though changing the gradient to mirror that used in the actual purification (0 to 50% MeCN over 6 min), the results were much more comparable, Figure 4.
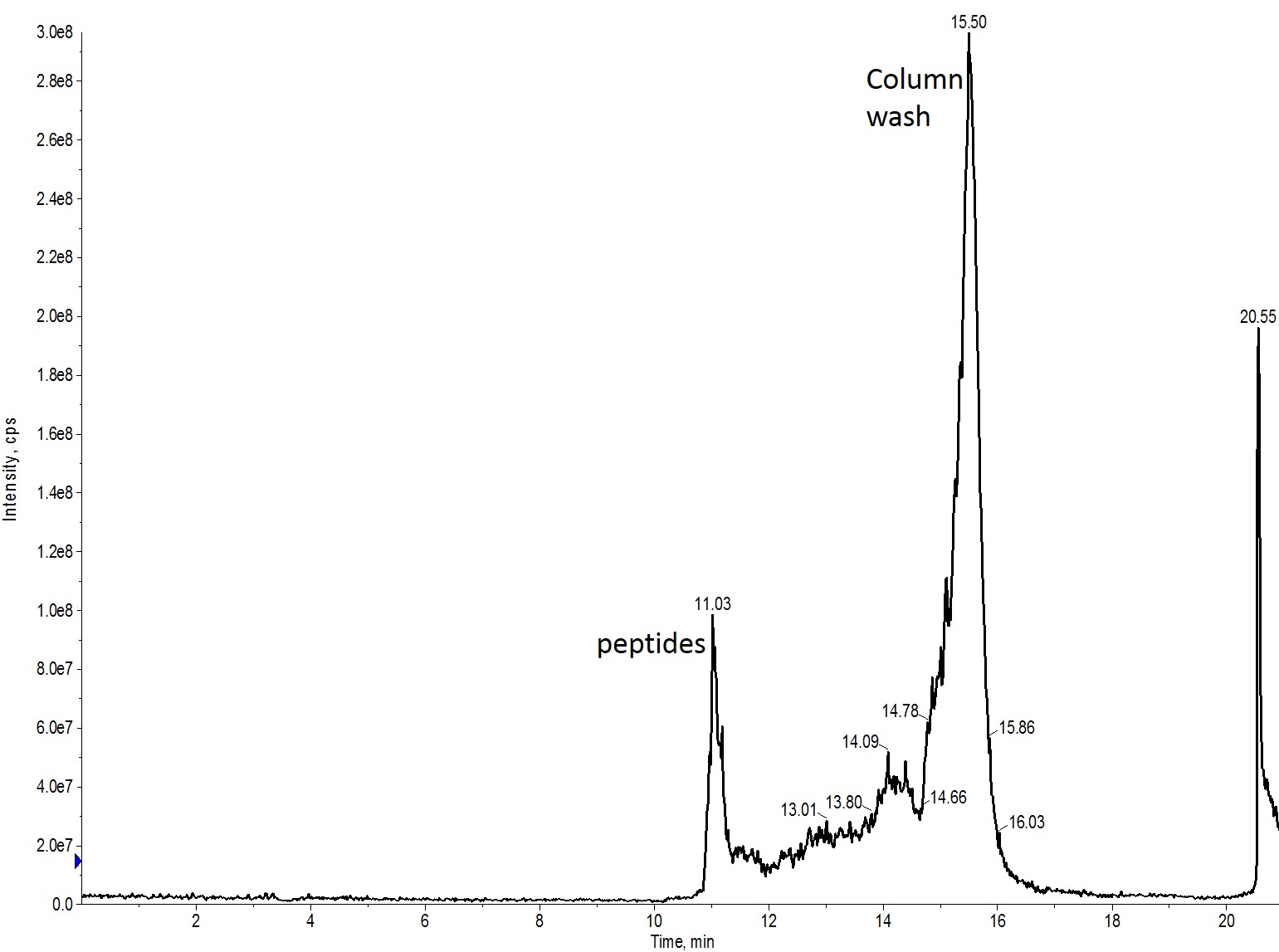
Figure 4: Crude peptide mixture evaluated using a BioC18 scout column. The sample was evaluated using a 0 to 50% MeCN gradient over 6 min. This evaluation is most similar to what is used with the flash purification system in both linear velocity as well as gradient slope.
This gradient is run at an 8.3% per minute slope, very fast when compared to more standard HPLC methods but has typically worked well for me as a starting point in my flash purification efforts. There is certainly room for some further optimization here. Importantly though, this provides a quick strategy for evaluating gradient purification efficiency without sacrificing a significant amount of the peptide sample.
I mentioned above the importance of matching gradients for consistent performance, but one thing that I have yet to mention is the importance of linear velocity when translating methods from one column/instrument to another. For the purification efficiency to be accurately recapitulated, the linear velocity MUST be held constant, or as close to it as possible. Just as in the above example where the gradient slope affected the resolution, so will linear velocity.
To learn more about strategies to increase your purification efficiency using flash chromatography, click the link below.